

Choroby rzadkie z definicji dotykają mniej niż 1 osobę na 2000, a liczba obecnie znanych nam chorób wynosi ok. 6-7 tysięcy. Wystarczy spojrzeć na dowolną stronę spisu chorób, aby uświadomić sobie jak niewiele o nich wiemy (dostępny tutaj).
Często pojawia się myśl: to mnie nie dotyczy, skala problemu jest niewielka. Między innymi z tego względu występują problemy z diagnozą. Mimo licznych przeciwności losu, osoby dotknięte chorobami rzadkimi cechują się siłą, jakiej mógłby im pozazdrościć niejeden zdrowy człowiek. Nie inaczej jest w przypadku dwóch wspaniałych osób, bohaterów dzisiejszego wywiadu – Przemka i Karoliny, których mieliśmy okazję lepiej poznać podczas XVII Międzynarodowej Konferencji Chorób Rzadkich, która w tym roku odbyła się pod hasłem „Nie przegap choroby rzadkiej”. Aby pomóc osobom dotkniętym rzadkimi schorzeniami, powstają stowarzyszenia i fundacje, których jednym z celów jest szerzenie wiedzy o chorobie. Takie przedsięwzięcie rozpoczęli nasi rozmówcy, taką działalność prowadzi także Stowarzyszenie Chorych na Mukopolisacharydozę i Choroby Rzadkie (MPS), które wspomnianą konferencję organizuje od 17 lat.
Choroba Pompego – czym jest?
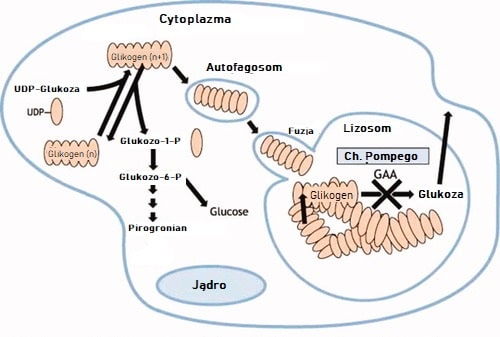
Choroba Pompego, zwana glikogenolizą typu II, należy do chorób spichrzeniowych glikogenu, w której to dochodzi do zmniejszonej aktywności lub całkowitego braku enzymu – kwaśnej maltazy (GAA, acid alpha glucosidase). Enzym ten odpowiada za rozkład zgromadzonego w lizosomach glikogenu do glukozy. Jego brak oznacza „przeładowanie” organellum wielocukrem, pękanie i niszczenie komórek, głównie mięśniowych. Choroba jest dziedziczona autosomalnie recesywnie [2,3]. Obecnie w Polsce jest około 50 chorych.
Wyróżniamy trzy postaci choroby: niemowlęcą, dziecięcą i dorosłą. W pierwszym przypadku aktywność enzymu jest najczęściej mniejsza niż 1% lub obserwuje się całkowitą jego nieobecność. Rokowania w postaci niemowlęcej nie są najlepsze – pacjent często nie dożywa 1-2 r.ż. Wystąpienie pierwszych objawów w późniejszym okresie życia te rokowania znacząco poprawia, aktywność enzymu również jest większa [3].
DIETETYCY.ORG.PL: Kiedy doszło u Ciebie do pierwszych objawów i jakie były wstępne diagnozy?
Przemysław Burmer: Pierwsze objawy w zasadzie dotyczyły aktywności i wydolności fizycznej. Zawsze byliśmy nieco słabsi od naszych rówieśników, co lekarze ogólni tłumaczyli tym, że byliśmy wcześniakami. Na lekcjach wf-u przybiegaliśmy ostatni. W czasie poborów do wojska w 1995 roku zacząłem chodzić od lekarza do lekarza, trafiłem w końcu do neurologa. Diagnoza – dystrofia mięśniowa i oczywiście kategoria D na komisji. W 2000 roku trafiłem do poradni neurologicznej, gdyż miała mi się urodzić pierwsza córka. U Karoliny zdiagnozowano wtedy dystrofię mięśniową Kugelberga-Welandera (postać młodzieńcza rdzeniowego zaniku mięśni, przyp. autora). Na podstawie wyników biopsji mięśniowej, gdzie wykazano m.in. bardzo podwyższony poziom kinazy kreatynowej, stwierdzono rozpad pierwotny mięśnia. Potwierdzono diagnozę dystrofii. Mój stan był wtedy jeszcze całkiem dobry, aż do pierwszego upadku, kiedy doszło do złamania kompresyjnego 12. kręgu piersiowego. Dostałem gorset Jewetta, długo pozostawałem bez ruchu i odbiło się to na sprawności moich mięśni. Później dochodziło do kolejnych złamań części lędźwiowej i zabiegu cementoplastyki. Do operacji poddano mnie całkowitej narkozie, co nie powinno mieć miejsca. Przyczyniło się to do osłabienia mięśni oddechowych i niewydolności, która w wyniku braku leczenia pogłębiała się. Po roku trafiłem do szpitala w wyniku nagłego zatrzymania krążenia (NZK), wprowadzono mnie w stan śpiączki farmakologicznej, w której pozostawałem 10 dni. Po miesiącu dopiero dostałem respirator i mogłem wrócić do domu.
Jakoś tak się złożyło, że trafiliśmy z siostrą do cenionego genetyka w Gdańsku, który zainteresował się wynikami naszych badań i uznał, że diagnoza mogła być źle postawiona. Po konsultacjach z innym lekarzem w Warszawie stwierdzono, że to choroba Pompego.

DIETETYCY.ORG.PL: Prawidłowa diagnoza została postawiona dopiero po ponad 10 latach, w 2011 roku?
Przemysław Burmer: Tak, a im wcześniej choroba zostaje rozpoznana, tym szybciej można wprowadzić terapię i hamować dalszą progresję. Niestety trzeba przyznać, że jest to dość trudne, ciężko mówić o typowych objawach, a po niektórych pacjentach w ogóle nie widać, że cierpią na Pompego. Pojawia się wzmożona męczliwość, osłabienie mięśni i duszności.
Charakterystyczne objawy i rozpoznanie
Objawy mogą różnić się u poszczególnych osób, ze względu na postać choroby, czy stopień aktywności enzymu. Jednakże można wyróżnić kilka charakterystycznych, do których należą:
- postępujący niedowład mięśni kończyn dolnych i obręczy biodrowej oraz mięśni stabilizujących,
- niewydolność oddechowa i znaczna męczliwość,
- w postaci dziecięcej: niewydolność serca [3,4].
Podstawą do rozpoznania choroby jest wynik badania suchej kropli krwi, w której określa się aktywność enzymu GAA. Wynik można potwierdzić także analizą próbki z krwi obwodowej. Dodatkowo w chorobie obserwuje się podwyższone stężenie kinazy kreatynowej (CK) i enzymów wątrobowych (AlAT i AspAT).
Leczenie
Leczenie w chorobie Pompego polega na enzymatycznej terapii zastępczej. Brakujący enzym, pozyskiwany z mleka transgenicznych chomików, dostarcza się w formie kroplówki w szpitalach co 2 tygodnie. Terapia w Polsce jest refundowana, co czyni ten przypadek jedyną chorobą nerwowo-mięśniową, której leczenie w naszym kraju jest refundowane [6]. Dawka leku wynosi 20 mg/kg m.c.

DIETETYCY.ORG.PL : Czy wiecie, jako którzy w Polsce zostaliście zdiagnozowani?
Przemysław Burmer: W tamtej chwili było nas wszystkich około dwudziestu osób.
DIETETYCY.ORG.PL: Terapia została włączona od razu?
Przemysław Burmer: Tak, ale w programie charytatywnym, gdyż refundacja była wówczas „wyłączona” z powodu nieefektywności kosztowej u osób z postacią dorosłą choroby, jednak dzięki staraniom społeczności chorych przywrócono finansowanie.
DIETETYCY.ORG.PL: Jak wygląda terapia enzymatyczna?
Przemysław Burmer: Pozyskiwany lek, Myozyme, pełną aktywność nabiera dopiero po wniknięciu do lizosomów. U niemowląt, gdzie endogenny enzym praktycznie nie jest produkowany, do takiej aktywacji zazwyczaj nie dochodzi, często rodzicom nawet się nie mówi o rozpoznaniu choroby u dziecka, gdyż terapia nie będzie skuteczna. Minimalna aktywność enzymu u niemowląt stwarza pewne szanse na wydłużenie czasu życia. Ostatnio zmarła dziewczynka z postacią niemowlęcą, która miała taką niewielką aktywność i dożyła 5 lat na leczeniu.
My dostajemy co 2 tygodnie kroplówkę z lekiem, dawka jest wyliczana indywidualnie, w oparciu o masę ciała. Podanie leku musi odbywać się w warunkach szpitalnych. Oprócz tego co pół roku jesteśmy badani, celem określenia skuteczności terapii.
DIETETYCY.ORG.PL: Jak wyglądają te badania okresowe?
Przemysław Burmer: Co pół roku pojawia się zdenerwowanie, bo badania mogą wykazać, że enzym nie działa jak powinien, że nie ma efektu i terapia zostaje wtedy przerwana. Bada się między innymi stężenie kinazy kreatynowej, enzymów wątrobowych, ma miejsce badanie dna oka i słuchu, badania kardiologiczne, RTG, test marszu, spirometria, dodatkowo konsultacje ortopedyczne, internistyczne i inne.
DIETETYCY.ORG.PL: Karolino, jesteś pierwszą osobą w Polsce i trzecią na świecie, która otrzymywała enzym będąc w ciąży. Czy terapia była prowadzona od początku jej trwania?
Karolina Wojtacha: Nie, to była walka. W pierwszym miesiącu, wiedząc już, że jestem w ciąży, zgłosiłam to i terapia automatycznie została mi zabrana. Wynikało to z faktu, że w wytycznych dotyczących leku widnieje informacja o zakazie terapii w przypadku ciąży i laktacji. Dotarliśmy do badań klinicznych z innych krajów, dotyczących przebiegu ciąży u dwóch kobiet, które otrzymywały enzym i wywalczyliśmy od trzeciego miesiąca charytatywną terapię u producenta leku. Charytatywnie, bo NFZ opierał się na zapisach i ustawach i wspomniane badania nie mogły tego zmienić. Po urodzeniu dziecka i podpisaniu wielu dokumentów zostałam przywrócona do programu terapii. Jednym z warunków była rezygnacja z karmienia dziecka.

DIETETYCY.ORG.PL: Jak przebiegała ciąża, jak się czułaś i czy były jakieś powikłania?
Karolina Wojtacha: Ciąża przebiegła bez żadnych komplikacji, czułam się dobrze. Jakiekolwiek występujące objawy były związane jedynie z ciążą. Jedynie pogorszyły mi się wyniki badań przez ten okres bez terapii. Chociaż może to wydawać się śmieszne, to lepiej czułam się będąc w ciąży.
Przemysław Burmer: Mieliśmy ostatnio podobną sytuację, bo inna kobieta z chorobą Pompego zaszła w ciążę. Była w dużo lepszym stanie, bo otrzymywała terapię enzymatyczną od bodajże 7 r.ż., ale decyzja znowu była ta sama: wyłączenie z programu. Jeden z lekarzy profesorów podjął się prowadzenia tej osoby, bo zna sytuację, ma wyniki badań Karoliny. Fundacja też pisała, aby nie wyłączać Jej z programu, wstawiła się za Nią także jedna Pani Poseł, członkini Komisji Zdrowia. Ponownie było możliwe jedynie finansowanie charytatywne, ale tym razem nie wyrazili zgody prawnicy szpitala, bo „ryzyko było zbyt duże”, chociaż dziewczyna była w dużo lepszym stanie, niż Karolina, kiedy zaszła w ciążę. Chcemy teraz wyjaśnić całą sprawę.
DIETETYCY.ORG.PL: A jak przebiegł sam poród?
Karolina Wojtacha: Musiało być przeprowadzone cesarskie cięcie, gdyż poród siłami naturalnymi jest zbyt dużym obciążeniem dla organizmu. Osoby z chorobą Pompego cierpią na nietolerancję wysiłku.
Zmiany w organizmie chorego i rehabilitacja
W wyniku choroby w organizmie pacjenta dochodzi do szeregu zmian w funkcjonowaniu nie tylko mięśni, ale wielu innych układów i narządów, m.in. w:
- sercu – złaszcza w postaci niemowlęcej obserwuje się przerost serca i postępującą dysfunkcję;
- płucach – w miarę postępu choroby Pompego mięśnie oddechowe osłabiają się, co prowadzi do niskiej objętości płuc, upośledzonego kaszlu, zaburzeń gazometrii i zaburzeń oddychania podczas snu;
- układzie pokarmowym – osłabienie mięśni może prowadzić do zaburzeń żucia i przełykania (dysfagii), zwolnionej perystaltyki jelit;
- kościach – u chorych obserwuje się osteopenię i osteoporozę;
- układzie nerwowym – może dochodzić do akumulacji glikogenu w mózgu, jądrach pnia mózgu i innych komórkach nerwowych, mogą pojawić się zmiany w nerwie wzrokowym, utrata słuchu [3].
Aby opóźniać progresję wymienionych zaburzeń i w porę reagować, potrzebne są okresowe badania. Z tego względu co pół roku, w czasie kontroli efektów leczenia enzymem, wykonuje się również badania serca, spirometrię, DEXA, badanie słuchu czy wzroku. Ważna jest również konsultacja lekarska i dostosowana do pacjenta rehabilitacja. Niektóre badania podają, że niewielki wysiłek aerobowy w połączeniu z terapią enzymatyczną może poprawiać stan pacjenta i przeciwdziałać rozpadowi mięśni czy osteoporozie. Nie u wszystkich jednak jego wprowadzenie jest możliwe. Istotna jest także rehabilitacja oddechowa, ćwiczenia rozciągające i stabilizujące. Pod żadnym pozorem nie można pacjentowi zalecać ćwiczeń siłowych, należy unikać wzmożonego wysiłku [3,7].

DIETETYCY.ORG.PL: Jak wygląda u Was rehabilitacja?
Przemysław Burmer: Przede wszystkim nie ma możliwości aby wprowadzić u nas jakiś większy wysiłek. Może być on możliwy u innych, ale zdecydowanie pod kontrolą, właśnie ze względu na znaczne osłabienie mięśni nóg. Z racji faktu, że zostaliśmy włączeni do programu respiratoterapii, dwa razy w tygodniu przysługuje nam rehabilitant. Wykonujemy ćwiczenia oddechowe.
DIETETYCY.ORG.PL: W Twoim przypadku do znacznej niewydolności oddechowej doszło po zabiegu cementoplastyki w znieczuleniu ogólnym, a u Ciebie Karolina?
Karolina Wojtacha: Mi również podano znieczulenie ogólne podczas cesarskiego cięcia przy rozwiązaniu drugiej ciąży. Były problemy z wybudzeniem mnie. Potem pojawiła się niewydolność oddechowa i spowodowane nią niedotlenienie: czułam się zmęczona, miałam częste bóle głowy.
Przemysław Burmer: Też warto tutaj wspomnieć o kolejnym absurdzie: żeby włączyć pacjenta do respiratoterapii, wniosek musi wyjść z OIOMu. To znaczy, że stan pacjenta musi pogorszyć się na tyle, żeby konieczne było położenie go na oddziale intensywnej terapii. Karolina chodziła do pulmonologa, wyniki badań wskazywały na postępującą niewydolność, ale nic nie dało się zrobić. Często musi dojść do tragedii, żeby zostały podjęte pewne działania. Teraz walczymy o to, aby zapisy dotyczące respiratoterapii zmienić, bo niewydolność oddechowa wpisuje się niejako w chorobę Pompego.

DIETETYCY.ORG.PL: Czy słyszeliście kiedyś o diecie w chorobie Pompego lub czy otrzymaliście jakiekolwiek zalecenia?
Przemysław Burmer: Zalecenia są raczej ogólnikowe. Wiemy, że mamy jeść posiłki lekkostrawne, mniejsze porcje, ale częściej, ze względu na męczliwość i problemy z oddychaniem. Mówiono także o większej ilości spożywanego białka. Sam wiem po sobie, że jak jem za dużo, to kończę na stojąco, żeby zmniejszyć nacisk na przeponę.
Karolina Wojtacha: Do tego mamy także zapobiegać nadwadze, uprawiać aktywność fizyczną w miarę możliwości.
Dieta w chorobie Pompego
Zaleceń żywieniowych rzeczywiście jest mało, a jeszcze mniej badań, które potwierdzałyby ich działanie. Pacjentom zaleca się dietę niskowęglowodanową i wysokobiałkową, o proporcjach 30-35% podaży energii z węglowodanów (ale nie mnie niż 130-150 g), 20-25% z białek i 35-40% z tłuszczów. Może to zapobiegać powstawaniu nadmiernych ilości glikogenu i chronić mięśnie przed rozpadem. Ponieważ do pozyskania energii z tłuszczów potrzeba mniej tlenu, dieta wysokotłuszczowa może być pomocna u osób z niewydolnością oddechową. Z drugiej jednak strony duża ilość tego składnika obciąża przewód pokarmowy, dlatego wskazane jest jedzenie nawet 8-10 mniejszych posiłków w ciągu dnia [2,7,8,9].
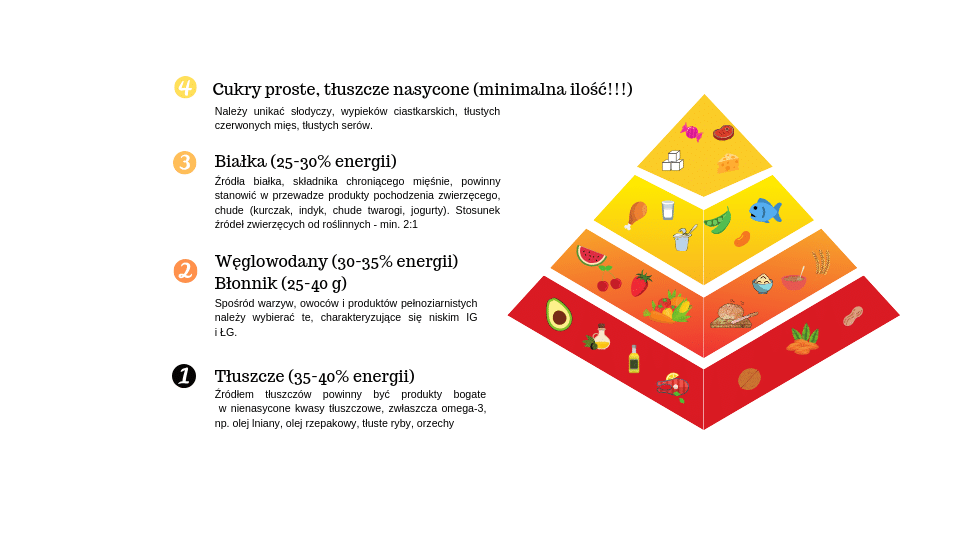
Źródłami tłuszczów powinny być przede wszystkim oleje roślinne (lniany, rzepakowy, oliwa z oliwek), tłuste ryby, orzechy i nasiona. Pacjenci powinni wybierać produkty zawierające węglowodany złożone, a białko dostarczać wraz z mięsem czy nabiałem, aby było ono jak najlepiej przyswajalne [2,9]
W przypadku problemów z żuciem i połykaniem należy podawać pokarmy w formie płynnej lub pół-płynnej. Aby zapobiegać zakrztuszeniom można zagęszczać wodniste potrawy. Zależnie od tego, czy występują zaparcia, czy biegunki, trzeba modyfikować podaż błonnika w diecie [10].
W żywieniu należy również zadbać o odpowiednią podaż wszystkich witamin i minerałów, zwłaszcza wapnia i witaminy D3 oraz kontrolować stan odżywienia organizmu [2].
Pacjent zdecydowanie musi zrezygnować z alkoholu i palenia papierosów.
DIETETYCY.ORG.PL: Założyliście wraz z Pawłem Janikiem fundację chorób rzadkich, Fundację Rare Diseases. Gdybyś mógł o niej krótko opowiedzieć.
Przemysław Burmer: Inspiracją były wszystkie nasze przeżycia. Od kiedy dowiedziałem się o chorobie, zacząłem więcej się dowiadywać, poznawać innych chorych, mówimy na siebie „Pompki”. Lubię gadać, taki mam styl bycia, chciałem coś robić w tym kierunku, pomagać. Jak mnie drzwiami wypchną, to wejdę oknem {śmiech}. Zacząłem pytać, rozmawiać z innymi pacjentami o przeżyciach, problemach systemowych, małej świadomości lekarzy. Nie każdy potrafi wyrazić u lekarza swoje zdanie, też często lekarze nie potrafią słuchać, a później są tego konsekwencje. Gdy doszło u mnie do złamania podkrętarzowego kości ok. 3 lata temu, operacja (zespolenie płytkami, przyp. red.) przebiegła bez komplikacji, bo przypadek konsultował anestezjolog, który się na tym znał. Przy wyjmowaniu „blachy” zabrakło już wiedzy i podano mi zbyt dużą dawkę znieczulenia, co nieomal zakończyłoby się moją śmiercią na skutek kolejnego NZK, gdyby nie przytomność sąsiada z sali. Nawet odpowiednia procedura po operacji, czyli usadowienie mnie w pozycji pół-siedzącej, mogłaby zapobiec powikłaniom, mimo za dużej dawki. Zamiast tego leżałem i znieczulenie „poszło” do serca. Wtedy lekarze zawiedli po całości.
Ponieważ w pojedynkę niewiele można zdziałać, postanowiłem zarejestrować fundację i działać na każdej płaszczyźnie, na której się da, podnosić świadomość służby zdrowia, rodzin i bliskich osób chorych, wspierać ich w trudnych momentach i walczyć dla nich o lepsze jutro.
DIETETYCY.ORG.PL: Jakie są plany na przyszłość?
Przemysław Burmer: Chcemy dalej rozwijać fundację i zapewnić każdemu choremu należytą, godną opiekę zdrowotną, począwszy od diagnozy, przez leczenie, a na rehabilitacji skończywszy. Jesteśmy w trakcie tworzenia strony internetowej, która docelowo będzie poruszać temat nie tylko choroby Pompego, ale też innych chorób rzadkich. Będzie to swego rodzaju encyklopedia, zbiór informacji o chorobie, opis charakterystycznych symptomów, pomocnych w diagnozie, dieta. To wydają się być małe rzeczy, ale wszystkie razem będą naprawdę pomocne. Mamy nadzieję, że stronę uda się ukończyć w niedługim czasie, ale jednocześnie chcemy, aby to było naprawdę coś. Praca osób w fundacji ma charakter wolontaryjny, tym bardziej należy się im uznanie. Staramy się o różnego rodzaju granty, bo znacznie przyspieszy to prace. Patrzymy z optymizmem w przyszłość i chcemy, aby była ona też optymistyczna dla wszystkich dotkniętych chorobami rzadkimi.
Polecane strony:
https://www.uniqius.org/ – strona Fundacji Rare Diseases (w moderacji)
http://chorobyrzadkie.pl/ – strona stowarzyszenia MPS
https://www.worldpompe.org/ – strona poświęcona chorobie (w języku angielskim)
https://www.orpha.net/consor/cgi-bin/index.php – baza danych o chorobach rzadkich
https://www.eurordis.org/ – pozarządowa, europejska organizacja pacjentów z chorobami rzadkimi
Źródła:
- http://www.orpha.net/national/PL-PL/index/co-to-jest-choroba-rzadka/
- Cupler, E. J., Berger, K. I., Leshner, R. T., Wolfe, G. I., Han, J. J., Barohn, R. J., … & AANEM CONSENSUS COMMITTEE ON LATE‐ONSET POMPE DISEASE. (2012). Consensus treatment recommendations for late‐onset Pompe disease. Muscle & nerve, 45(3), 319-333.
- Kishnani PS, Steiner RD, Bali D, et al. Pompe disease diagnosis and management guideline. Genet Med. 2006;8(5):267-288
- Hirschhorn R, Reuser AJ. Glycogen storage disease type II: acid α-glucosidase (acid maltase) deficiency. In: Scriver CR, Beaudet AL, Valle D, Sly WS, eds. The Metabolic & Molecular Bases of Inherited Disease. 8th ed. New York, NY: McGraw-Hill; 2001:3389-3420.
- Goldstein JL i wsp. Screening for Pompe disease using a rapid dried blood spot method: experience of a clinical diagnostic laboratory. Muscle Nerve. 2009;40(1):32-36.
- http://chorobyspichrzeniowe.pl/choroba-pompego/
- Slonim, A. E., Bulone, L., Goldberg, T., Minikes, J., Slonim, E., Galanko, J., & Martiniuk, F. (2007). Modification of the natural history of adult‐onset acid maltase deficiency by nutrition and exercise therapy. Muscle & nerve, 35(1), 70-77.
- Heller, S., Worona, L., & Consuelo, A. (2008). Nutritional therapy for glycogen storage diseases. Journal of pediatric gastroenterology and nutrition, 47, S15-S21.
- Esposito, K., Imrota, M.R., Giugliano, D. (2011). The Nutritional Approach to Pompe Disease. Acta Myologica, 30(3): 208–209.
- Nutrition and Dietary Therapy, worldpompe.org, 2005



{kind=link}
{kind=link}
{kind=link}