

Zespół Pradera-Williego został po raz pierwszy opisany przez Johna Down’a w drugiej połowie XIX wieku [3]. Do dziś jest jednym z najpoważniejszych zagrożeń dla zdrowia i życia. Jej wykrycie wiąże się z wielką rewolucją w całej rodzinie. Narastająca w szybkim tempie masa ciała prowadzi bowiem do wielu komplikacji. Czym jest dokładnie ten zespół i czy możliwe jest jego wyleczenie?
Czym jest zespół Pradera-Williego
Zespół Pradera-Williego (PWS) często nazywany jest chorobą wiecznego głodu. Jest to schorzenie uwarunkowane genetycznie. Dotąd nie wynaleziono żadnego leku, który przeciwdziałałby objawom choroby. Zespół ten powstaje w wyniku poważnych zmian w układzie genów znajdujących się na chromosomie 15. Obecnie poznane są trzy rodzaje błędów genetycznych, które odpowiadają za objawy tego zespołu. Zalicza się do nich:
- delecje fragmentu chromosomu 15 pochodzącego od ojca, obejmująca region delecja 15q11-13 (około 70% przypadków);
- disomia matczyna, czyli dziedziczenie dwóch chromosomów 15 od matki (około 25% przypadków);
- mutacja centrum imprintingu – odojcowskie geny są obecne w regionie 15q11-13, ale są nieaktywne. W wyniku czego dochodzi do nieprawidłowego rozwoju (około 5%) [1, 2].
Fazy żywieniowe
Charakterystyczną cechą Zespołu Pradera-Williego są zaburzenia odżywiania. U każdego pacjenta mogą przebiegać z różnym nasileniem [3]. Określono kilka faz żywieniowych. Wyróżnia się pięć głównych faz oraz podgrupy do fazy pierwszej i drugiej. Faza zerowa odnosi się już do życia płodowego. Cechuje się on słabymi ruchami płodu oraz ograniczeniem wzrastania.
Faza pierwsza
W fazie 1 (0-9 miesięcy) noworodki wykazują słabe napięcie mięśniowe. Występuje słabe ssanie mleka matki, co wiąże się z niewielkim spożyciem pokarmu. W konsekwencji często wymagane jest żywienie z zastosowaniem zgłębnika. Czasami polecane jest stosowanie specjalnych sutków do karmienia. Jest to niezbędne do utrzymania prawidłowej masy ciała.
Natomiast w okresie 9-25 miesięcy (faza 1b) poprawia się apetyt. Zazwyczaj nie jest wymagane karmienie wspomagane. Obserwuje się także prawidłowy wzrost.
Faza druga
Faza 2 odznacza się przyrostem masy ciała dziecka. Wyróżniono tutaj dwie podfazy. W fazie 2a (od około 2 do 4,5 lat) występuje wzrost masy ciała, jednakże nie ma znaczących zmian w spożyciu pokarmu. Z kolei w fazie 2b (4,5-8 lat) przyrost masy ciała wiąże się z jednocześnie zwiększonym zainteresowaniem żywnością.
Faza trzecia i czwarta
Faza 3 wyróżnia się hiperfagią. Zazwyczaj towarzyszy jej poszukiwanie żywności i brak uczucia sytości. Nie każda osoba z PWS musi przejść przez te wszystkie fazy. Poza tym niektóre osoby mogą przejść do fazy 4. Oznacza to, że potrafią zapanować nad swoim apetytem oraz odczuwają sytość po spożyciu posiłku [4, 5, 6]. Charakterystyczne cech faz żywieniowych zostały podsumowane w tabeli 1.
Tabela1. Fazy żywieniowe w zespole Pradera-Williego.

Charakterystyczne cechy i objawy
Hipotonia
Obniżone napięcie mięśniowe (hipotonia) jest pierwszym charakterystycznym objawem PWS. W początkowym okresie wykazuje wysokie nasilenie. Z wiekem stopniowo ustępuje. Hipotoniczne niemowlę cechuje wiotkość:
- w pozycji na brzuchu nie jest w stanie podnieść główki;
- mało się rusza;
- mało płacze;
- przy braniu na ręce sprawia wrażenie bezwładnego [1].
Charakterystyczny jest również słaby odruch ssania. Podczas karmienia występują trudności z połykaniem. Czasami dzieci szybko męczą się podczas jedzenia. Niektóre dzieci z PWS mogą wykazywać brak odruchu ssania. Przyczynia się to do trudności w karmieniu. W efekcie dzieci narażone są na niedobory pokarmowe i energetyczne. Dlatego też w tym okresie dziecko cechuje niedowaga [1].
Hiperfagia i brak uczucia sytości
Hiperfagia objawia się niepohamowanym apetytem i zaburzeniem odczuwania sytości. Jest to cecha, która wyróżnia ten zespół spośród innych chorób. Objaw ten przepisuję się dysfunkcji podwzgórzowo-przysadkowej.
Stwierdzono, że u osób z PWS występuje podwyższone stężenie greliny. Hormonu odpowiedzialnego za stymulację głodu i promowanie przyjmowania pożywienia [3]. Zazwyczaj zaczyna pojawiać się między 2. a 6. rokiem życia. Do tej pory dziecko, które miało trudności z jedzeniem, teraz wykazuje nadmierne nim zainteresowanie.
Podane posiłki zjada w całości. Rzadko zostawia resztki na talerzu. Poszukuje także dodatkowych porcji. Objaw hiperfagii może być bardzo niebezpieczny. Wynika to z faktu, że niektóre dzieci mogą być w stanie zjeść wszystko. Np. substancje niejadalne, a nawet trujące (środki czystości, zepsute jedzenie wyciągnięte z kosza na śmieci) [1]. Brak odczuwania sytości i nadmierna konsumpcja żywności prowadzi do przyrostu masy ciała. W konsekwencji dochodzi do rozwoju otyłości. Wiąże się to z wieloma zaburzeniami stanu zdrowia.
Obniżona przemiana materii
U osób z PWS obserwuje się obniżoną przemianę materii. Jest to bardzo niekorzystne, gdyż dodatkowo sprzyja rozwojowi otyłości. Badania wskazują, że u tej grupy osób występują niższe spoczynkowe wydatki energetyczne. Niektóre badania wykazują także na obniżenie termogenezy indukowanej pożywieniem u osób z PWS [5]. W tym przypadku niezbędne jest obniżenie wartości energetycznej diety oraz włączenie aktywności fizycznej. Wpłynie to na poprawę przemiany materii. Brak podjęcia odpowiednich działań przyczyni się do szybkiego przyrostu masy ciała.
Otyłość
Otyłość zaczyna rozwijać się po okresie trudności w karmieniu dziecka (faza 2). Jest to zazwyczaj otyłość typu centralnego (brzuch, uda, pośladki); [4]. Wynika ona z opisanych wcześniej zaburzeń, które towarzyszą temu zespołowi. Otyłość u osób z PWS różni się od otyłości ,,zwykłej”. Przede wszystkim tempo jej narastania w PWS jest bardzo szybkie. Poza tym przyrostowi masy ciała nie towarzyszy wzrost tkanki mięśniowej. Natomiast u osób bez PWS nadmierna konsumpcja zwiększa ilość tkanki tłuszczowej, ale i również masy mięśniowej [1].
Otyłość ma bardzo negatywny wpływ na stan zdrowia. Może prowadzić do wad postawy (koślawość kolan, skrzywienie kręgosłupa). Jest również wynikiem poważnych konsekwencji zdrowotnych, np. zwiększa ryzyko cukrzycy, chorób serca, układu oddechowego itp. Jakakolwiek aktywność fizyczna staje się coraz trudniejsza. Ograniczona ruchowość dodatkowo sprzyja przyrostowi masy ciała. Uważa się, że otyłość i związane z nią zaburzenia stanu zdrowia, są głównymi przyczynami przedwczesnej śmiertelności.
Niedobór hormonu wzrostu
Niedobór hormonu wzrostu (GH) jest typowym objawem PWS. Odpowiada ona za prawidłowy wzrost dziecka. Jego niedobór prowadzi zatem do zaburzeń wzrastania. Niski wzrost dziecka z PWS jest więc także bardzo charakterystyczną cechą. Ponadto hormon wzrostu jest niezbędny do prawidłowego wykształcenia mięśni. Obniżony poziom GH wpływa na występowanie zbyt małej masy mięśniowej. Niedobór tkanki mięśniowej to jedna z przyczyn obniżonego zapotrzebowania energetycznego [1, 3, 4].
Hipogonadyzm
Hipogonadyzm to nieprawidłowe funkcjonowanie układu rozrodczego. Objawia się np. hipoplazją narządów płciowych, zaburzeniami w rozwoju dojrzewania czy niepłodnością. U chłopców często obserwuje się małe prącie oraz niedorozwój moszny. Z kolei u dziewcząt mogą występować małe wargi sromowe lub niedorozwój łechtaczki [1, 4, 5].
W okresie dojrzewania u chłopców zazwyczaj nie pojawia się zarost. Rzadko również przechodzą mutację. U dziewcząt nie pojawia się miesiączka albo występuje ona nieregularnie. Stopień nasilenia hipogonadyzmu jest różny. Np. w niektórych przypadkach okres dojrzewania może być nieobecny, a w innych opóźniony. Do tej pory przyczyna występowania tego schorzenia u osób z PWS jest niejasna [5].
Rozwój psychoruchowy
U dzieci z PWS obserwuje się opóźniony rozwój psychomotoryczny. Występuje on u 90-100% osób z tym zespołem [4]. Już we wczesnym okresie można zauważyć opóźnienie rozwoju psychomotorycznego. Dzieci z tym zespołem zazwyczaj później nabywają umiejętność samodzielnego siedzenia czy chodzenia. Występuje także zaburzony i opóźniony rozwój mowy. Dzieci mają trudność z wypowiadaniem słów. Ich mowa jest niewyraźna, uproszczona, często niegramatyczna. Zaburzenia rozwoju psychomotorycznego bardzo wyraźne są w okresie szkolnym. Dzieci wykazują trudności w nauce. Mają problemy z abstrakcyjnym myśleniem. Zazwyczaj dobrze czytają, ale pisanie sprawia im wiele trudności [1].
Zaburzenia zachowania
Dzieci z zespołem Pradera-Williego cechuje bardzo emocjonalna postawa. W początkowym okresie życia zwykle wykazują takie cechy jak: przyjaźń, życzliwość, posłuszeństwo itp. Natomiast z wiekiem nasilają się cechy zupełnie przeciwne. Dzieci są drażliwe, skłonne do napadów złości, nieraz nawet popadające w agresję. Utrudnia to prawidłowe kontakty w społeczeństwie [1, 4].
Problemy behawioralne i psychiczne najbardziej zakłócają jakość życia w okresie dojrzewania i dorosłości. Starsi nastolatkowie i dorośli mogą wykazywać smutek, niepokój i depresję. Prowadzi to do negatywnego wizerunku i wycofania. Ponadto dzieci i młodzież mogą rozwinąć formę zaburzeń obsesyjno-kompulsywnych [5]. Charakterystyczna obsesja objadania się powoduje, że są w stanie zjeść śmieci czy produkty spożywcze zamrożone. Dopuszczają się także kradzieży.
Niektóre dzieci z PWS (około 30%) mają skłonność do obsesyjnego rozdrapywania i skubania skóry. Może to skutkować powstawaniem licznych, trudno gojących się ran. U tych osób występuje wysoki próg bólu. Według badaczy u chorych na PWS nie występują przykre doznania związane z tym zachowaniem. Odczuwają natomiast przyjemność. Wynika to prawdopodobnie z faktu, że ich mózg produkuje substancję uśmierzającą ból [1]. Takie zachowania są bardzo niebezpieczne. Grozi bowiem zakażeniami, a poza tym okalecza skórę.
Zachorowalność i śmiertelność
Zespół Pradera-Williego występuje u obu płci z taką samą częstotliwością: 1:10000–1: 30000 żywych urodzeń [2, 7]. Śmiertelność u osób z PWS jest wyższa w porównaniu do osób z niepełnosprawnością intelektualną. A główną jej przyczyną jest otyłość oraz wszelkie związane z nią powikłania stanu zdrowia. Na podstawie badania populacji, wskaźnik zgonów oszacowano na 3% rocznie.
Choroby układu oddechowego oraz sercowo-naczyniowego uważane są za najczęstsze przyczyny zgonów. Także cukrzyca, zakrzepowe zapalenie żył oraz zaburzenia ze strony przewodu pokarmowego uważane są za jedne z przyczyn zgonów [4]. Zespół Pradera-Williego jest złożonym stanem chorobowym. Niepohamowany apetyt prowadzi do rozwoju otyłości. Z kolei nadmierna masa ciała wiąże się z wieloma zaburzeniami ze strony różnych układów organizmu. Zaburzenie pracy wielu ważnych narządów i układów np. serca, układu krążeniowego czy oddechowego odpowiadają za śmiertelność osób z tym zespołem.
Diagnostyka
Kryteria rozpoznawania
Rozpoznanie zespołu Pradera-Williego najczęściej polega na ocenie cech fenotypowych, które składają się na ten zespół. W tym celu wykorzystuje się wykaz kryteriów (objawów) dużych (za 1 punkt) oraz małych (za 0,5 punktu). Został on opracowany przez Holm i wsp. (tabela 2); [4].
Tabela 2. Kryteria rozpoznania Zespołu Pradera-Williego.
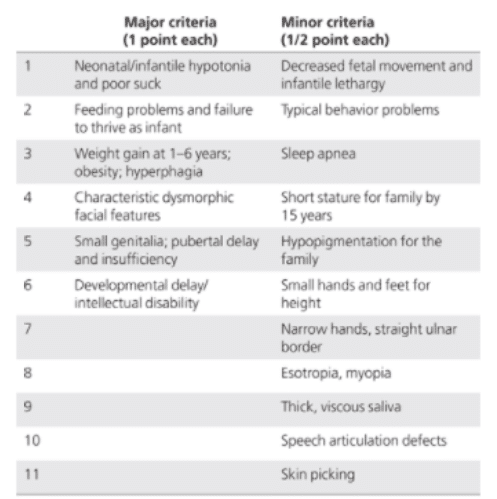
Diagnoza kliniczna wymaga pięciu punktów (co najmniej cztery z nich główne) w wieku poniżej trzech lat lub osiem punktów (co najmniej pięć z nich głównych) w wieku powyżej trzech lat.
Badania cytogenetyczne
Do oceny regionu krytycznego PWS wykorzystuje się badania chromosomów. Najczęściej wykonywane są metodami GTG, RBG, CBG o wysokiej rozdzielczości. Celem jest poszukiwanie utraty prążka 15q12 na jednym z chromosomów. Aby wykryć istnienie delecji fragmentu chromosomu 15, konieczna jest dalsza diagnostyka z użyciem metod cytogenetyki molekularnej (FISH). Pozwala ona na zidentyfikowanie utraty sygnału w obrębie regionu chromosomowego 15(q11-q13) [8].
Testy genetyczne molekularne
Zmiana wzoru metylacji DNA jest jednym z zjawisk warunkujących rozpoznanie PWS. Do stwierdzenia tej zmiany wykorzystuje się test metylacji DNA (SNRPN lub PW71B) z wykorzystaniem techniki PCR na podstawie oceny polimorfizmu długości fragmentów restrykcyjnych RFLP. Dzięki temu możliwe jest ustalenie, czy zmiany metylacji DNA są wynikiem mikrodelecji, czy disomii rodzicielskiej. Jeżeli diagnoza kliniczna nie zostanie potwierdzona, poszukuje się mutacji w genach centrum imprintingowego [8].

Suzanne B. Cassidy, et. al., Prader-Willi syndrome. str. 18
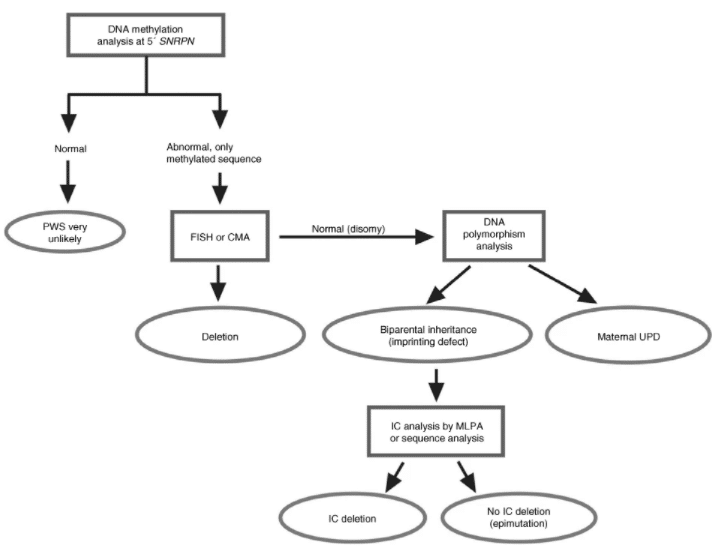
Schemat 1. Algorytm badań genetycznych dla zespołu Pradera-Williego (PWS).
Leczenie
Hormonalna terapia zastępcza
Suplementacja hormonem wzrostu jest jednym z elementów terapii zespołu PWS. Jest ona kluczowa dla przyrostu tkanki mięśniowej oraz wzrostu ciała. Przyczynia się także do przyspieszenia przemiany materii [1, 3]. Wzrost tkanki mięśniowej pozwala osobom z PWS na większą aktywność fizyczną. Ma tym samym korzystny wpływ na wykonywanie codziennych czynności.
Zwiększenie aktywności fizycznej przyczyni się do utrzymania należnej masy ciała. Poza tym wpływa pozytywnie na rozwój psychoruchowy. Terapia hormonalna powinna być rozpoczęta już od drugiego roku życia. Leczenie hormonem wzrostu jest bezpłatne do 18. roku życia. Jednakże wiele danych podaje, że terapia powinna być kontynuowana w wieku dorosłym. Ponieważ niedobór hormonu wzrostu utrzymuje się także w tym okresie. A to wpływa niekorzystnie na przemianę materii i aktywność [1].
Terapia zintegrowana
Leczenie osób z PWS wymaga zaangażowania specjalistów wielu dziedziny:
- lekarza — dzięki bogatemu doświadczeniu łatwiej i szybciej niż rodzice wychwyci występujące nieprawidłowości i skieruje dziecko do odpowiedniego specjalisty;
- endokrynologa – nadzoruje terapię hormonem wzrostu;
- dietetyka – układa plan żywienia oraz nadzoruje dietę. Dba o prawidłowe zbilansowanie żywienia osoby chorej na PWS. Kontroluje masę i skład ciała. Jest także wsparciem.
- psychologa – jest niezbędny w przypadku zaburzeń zachowania. Jest dobrym wsparciem dla osoby z PWS i jego bliskich;
- rehabilitanta lub fizjoterapeuty – pomaga sprawnie i szybko się rozwijać. Wpływa pozytywnie na przyrost masy mięśniowej. Zwiększa aktywność ruchową. Jest ważna dla utrzymania prawidłowej masy ciała.
- logopedy – terapia mowy i stymulacja rozwoju intelektualnego;
Proces leczenia składa się zatem z kilku form terapii. Nie będą one skuteczne, jeśli będą stosowane osobno. Ważne jest, aby specjaliści, pod których opieką chory się znajduje, w miarę możliwości ze sobą współpracowali.
Postępowanie żywieniowe: dieta w zespole Pradera-Williego
Dzieci w wieku do 4. roku życia
W pierwszych 6 miesiącach życia zaleca się karmienie piersią. Jeżeli nie jest to możliwe, należy stosować mieszanki mlekozastępcze. Do 9. miesiąca życia noworodki wykazują słaby apetyt. Utrudniony lub brak odruchu ssania prowadzi do spożycia niewielkiej ilości pokarmu. W konsekwencji niezbędne może być stosowanie specjalnych przyrządów lub technik podczas karmienia. W trudniejszych przypadkach konieczne jest karmienie z zastosowaniem kroplomierza lub strzykawki. Przypadki najcięższe mogą wymagać karmienia przez sondę. Prowadzone jest one w szpitalu pod kontrolą lekarza i dietetyka [1, 3].
Produkty uzupełniające należy wprowadzać około 5-6 miesiąca życia. Dieta pacjentów z PWS do 2. roku życia powinna być prawidłowo zbilansowana. Nie można stosować ograniczeń kalorycznych. W tym okresie następuje intensywny rozwój fizyczny i psychiczny dziecka.
Niezbędne jest więc dostarczenie mu wszystkich składników odżywczych w odpowiednich proporcjach. Przy wprowadzaniu nowych pokarmów należy wybierać produkty najbardziej wartościowe. Należy unikać włączania do żywienia takich produktów jak: słodycze, słone przekąski, napoje słodzone i gazowane itp.
Preferencje smakowe oraz nawyki żywieniowe kształtują się od wczesnego okresu. Dieta dziecka powinna zatem opierać się o takie produkty jak: mleko, jogurty naturalne, kefir, chudy twaróg, warzywa, owoce, chude mięso i przetwory mięsne oraz produkty zbożowe pełnoziarniste. Można włączać do diety nasiona roślin strączkowych, nasiona i orzechy. Te ostatnie, chociaż są wysokoenergetyczne, to jednak są dobrym źródłem błonnika i składników mineralnych.
Okres przedszkolny i szkolny
Osoby z PWS przez całe życie muszą stosować dietę niskoenergetyczną. Potrzebują oni zaledwie ½ porcji żywności wyznaczonej tabelami dla osób zdrowych. Jeżeli stosowana jest terapia hormonem wzrostu, zapotrzebowanie energetyczne zwiększa się do poziomu ¾.
Jeżeli nie będą przestrzegać diety, szybko rozwiną nadmierną masę ciała. W diecie należy zadbać o dostarczenie wszystkich niezbędnych składników odżywczych. Źródłem białka powinny być chude mięsa, ryby, chude przetwory mleczne oraz jaja.
Tłuszcze (np. masło czy oliwa) są niezbędne w niewielkiej ilości. Warto dodawać je do gotowych potraw np. łyżeczka oliwy do surówki. Odgrywają one ważną rolę m.in. w przyswajaniu witamin w nim rozpuszczalnych. Jako źródło węglowodanów należy wybierać pełnoziarniste produkty zbożowe. Mogą to być np. ciemne pieczywo, brązowy makaron czy ryż, płatki owsiane, kasze gruboziarniste. W diecie nie może zabraknąć również warzyw i owoców.
Uwagę należy zwrócić na różnego rodzaju dodatki. Stanowią one często źródło zbędnych kalorii. Nie należy stosować zasmażek, tłustej śmietany do zabielania. Przeciwwskazane jest także smażenie na głębokim tłuszczu itp.
Należy pamiętać, że sama dieta często nie wystarczy. Osoby z PWS nie są w stanie sami jej przestrzegać. Wykazują bowiem nienasycone łaknienie, dlatego wciąż poszukują okazji, aby coś zjeść. Dlatego tez bardzo ważna jest ich kontrola ze strony otoczenia. O chorobie dziecka warto poinformować pracowników przedszkola i szkoły. Należy przemyśleć sposób organizacji żywienia w stołówce czy podczas różnych wyjazdów, wycieczek. Warto być przygotowanym na to, że dziecko z czasem zacznie samo szukać jedzenia. Pomocne może okazać się zabezpieczenie pomieszczeń i szafek, w których znajduje się jedzenie – zamykanie kuchni, spiżarni na klucz [1]. Celem leczenia jest utrzymanie odpowiedniej masy ciała. Dążenie do poprawy zachowania właściwych proporcji składu ciała. Zmniejszenie zawartości tkanki tłuszczowej, a zwiększenie masy beztłuszczowej [3].
Okres dojrzewania i dorosłości
Ryzyko rozwoju nadmiernej masy ciała nadal jest wysokie w okresie dojrzewania oraz dorosłości. Osoby dorosłe z PWS mimo wielu lat terapii nadal nie potrafią zadbać o prawidłowe żywienie. Dlatego wciąż potrzebują oni kontroli ze strony innych osób.
Zapotrzebowanie energetyczne dla tych dwóch okresów życia nie zostało dokładnie określone. Uważa się jednak, że podaż kalorii powinna wynosić od 1000 do 1200 kcal dziennie. Dieta powinna być urozmaicona i dobrze zbilansowana. W ten sposób będzie dostarczać wszystkich niezbędnych składników odżywczych.

Niestety dieta ubogoenergetyczna jest trudna do zbilansowania. Niedobory w diecie najczęściej dotyczą witamin rozpuszczalnych w tłuszczach (A, D, E, K) oraz takich składników jak: wapń, żelazo i cynk. Warto także zwrócić uwagę na podaż kwasów omega-3. Ich dobry źródłem są głównie tłuste ryby morskie. Z uwagi na trudność w samodzielnym zbilansowaniu diety, warto zasięgnąć pomocy dietetyka. Ułoży on dietę dla pacjenta zgodnie z jego zapotrzebowaniem. Jest także dobrym wsparciem dla pacjenta oraz rodziny.
Jeżeli doszło do rozwoju nadmiernej masy ciała, niezbędna jest jej redukcja. Ogólnie dla tej grupy pacjentów zaleca się podaż 800-1000 kcal/dobę w przypadku redukcji masy ciała. Dieta powinna zawierać odpowiednie proporcje pomiędzy makroskładnikami np. 60% węglowodanów, 15% białek i 25% tłuszczy. W diecie należy uwzględniać węglowodany o niskim indeksie glikemicznym.
Podsumowanie
Zespół Pradera-Williego jest złożonym zespołem chorobowym uwarunkowanym genetycznie. Do tej pory nie ma żadnego leku, który przeciwdziałałby wszystkim objawom. Leczenie pacjentów z PWS nie jest łatwe i wymaga współpracy specjalistów z różnych dziedzin. Bardzo ważne jest wczesne rozpoznanie tego zespołu. Warunkuje to szybkie włączenie rehabilitacji, diety, czy leczenia hormonem wzrostu. Prawdziwy sukces w leczeniu dzieci z tym zespołem w ogromnej mierze zależy od zaangażowania, determinacji, poświęcenia ze strony ich rodziców, opiekunów, a nawet rówieśników. Przestrzeganie zasad terapii stanowi klucz do sukcesu w walce z chorobą.
Bibliografia:
- Libura M., Moje dziecko ma zespół pradera-williego. Jak mogę mu pomóc? Polskie Stowarzyszenie Pomocy Osobom z Zespołem Pradera-Williego, Warszawa 2007, str. 6, 12-14, 22-28, 39-42, 49, 50, 58, 62, 63, 67, 68,
- Kapczuk I., Beń-Skowronek I., Trojanowska-Szostek M., Kątska M., PraderWilli Syndrome – Diagnostics and Treatment. Endokrynologia Pediatryczna 2012, 11(3(40)), str. 81-88
- Krasińska A., Skowrońska B., Prader-Willi Syndrome – nutritional management in children, adolescents and adults. Pediatric Endocrinology Diabetes and Metabolism 2017, 23(2) – str. 101-106
- Suzanne B. Cassidy, MD , Stuart Schwartz, PhD , Jennifer L. Miller, MD and Daniel J. Driscoll, MD, PhD. Prader-Willi syndrome. Genetics in medicin 2012, 14(1) – str. 10-26
- Krystal A. Irizarry, MD, Mark Miller, MD, Michael Freemark, MD, Andrea M. Haqq, MD. Prader Willi Syndrome: genetics, metabolomics, hormonal function, and new approaches to therapy. Advances in Pediatrics 2016, 63(1), str. 1-33
- Miller L. J. Lynn H. Ch., Driscoll C. D., Nutritional Phases in Prader–Willi Syndrome. American Journal of Medical Genetics Part A 2011, 155A(5), str. 1-18
- Góralska M., Bednarczuk T., Rosłon M. i wsp. Management of Prader-Willi Syndrome (PWS) in adults — what an endocrinologist needs to know. Recommendations of the Polish Society of Endocrinology and the Polish Society of Paediatric Endocrinology and Diabetology. Endokrynologia Polska 2018, 69(4), str. 345-355
- Midro A. T., Olchowik B., Lebiedzińska A., Midro H., To know more about the Prader–Willi syndrome. Diagnosis. Psychiatria Polska 2009, 46(2), str. 135-150



{kind=link}
{kind=link}